Entendendo a MPS II - Síndrome de Hunter
A Mucopolissacaridose Tipo II (MPS II), também é conhecida como Síndrome de Hunter, é um distúrbio genético raro que afeta quase que exclusivamente indivíduos do sexo masculino.1

A Síndrome de Hunter pode afetar qualquer parte do corpo e causar diversos sinais e sintomas, sendo a combinação deles que pode indicar a doença. Na maioria das pessoas, os sinais surgem entre dois e quatro anos de idade e muitos vezes são queixas comuns da infância. Os sintomas mudam de pessoa para pessoa e, portanto, não há um padrão único típico.1

Entendendo a MPS II - Síndrome de Hunter
A Mucopolissacaridose Tipo II (MPS II), também é conhecida como Síndrome de Hunter, é um distúrbio genético raro que afeta quase que exclusivamente indivíduos do sexo masculino.1
A Síndrome de Hunter pode afetar qualquer parte do corpo e causar diversos sinais e sintomas, sendo a combinação deles que pode indicar a doença. Na maioria das pessoas, os sinais surgem entre dois e quatro anos de idade e muitos vezes são queixas comuns da infância. Os sintomas mudam de pessoa para pessoa e, portanto, não há um padrão único típico.1
Entenda as causas da Mucopolissacaridose Tipo II (MPS II)
A Síndrome de Hunter é uma das diversas doenças de depósito lisossômico (DDLs), que são condições onde as células não conseguem quebrar adequadamente certas moléculas. Há deficiência ou ausência total da enzima chamada iduronato-2-sulfatase (I2S), que em uma célula saudável quebra determinadas moléculas conhecidas como glicosaminoglicanos (GAGs). Antigamente, essas moléculas eram chamadas de mucopolissacarídeos, o que dá origem ao nome Mucopolissacaridose Tipo II.1,2
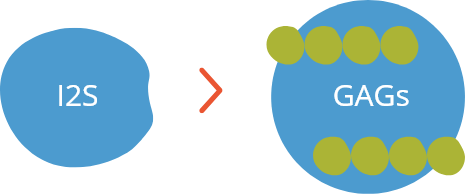
Se há deficiência ou ausência de I2S, os GAGs se acumulam e não deixam que a célula funcione de forma adequada.1 Todas as células saudáveis, exceto nos glóbulos vermelhos, produzem a enzima I2S e, então, sua deficiência pode afetar qualquer parte do corpo. É por isso que a MPS II pode ter sintomas em diversos órgãos e é chamada de doença multissistêmica.2